
ilus: 一个轻量级全基因组(WGS)和全外显子(WES)最佳实践分析流程生成器
不知觉间,距离我写下第一篇关于 WGS 数据分析系列的文章已经过去了三年多(WGS 系列文章),时间真的快啊。
很多朋友从当时的文章中得到了启发,对此我也很开心。在后来的日子里,我又合作完成了多个大规模的人类基因组学科研项目,在这个过程中关于大规模的 WGS 数据分析(数量从数千到十万、乃至百万级别)已经是家常便饭。
我自己也积累了更多属于大数据基因组学研究的分析策略、方法论和最佳实践,还在整理之中,现在把其中一部分做成 Python 工具包分享给大家。这个工具一年前写了第一个版本(当时只在我的知识星球里分享过一次),过程中也调试了碰到的问题,日趋完善,后续还将迭代更新。
这个工具我将其命名为 ilus (/i:ləs/),这是我看过的一部美剧——《无垠的太空》中通过星环抵达的第一个系外类地行星的名字。它是一个全面的、轻量的、可拓展且易用的半自动化全基因组(Whole genome sequencing, WGS)和全外显子(Whole exom sequencing,WES)分析流程生成器,是以前我 这篇文章 所提供代码的高级版本。
简介
ilus 的用途就是生成完整的 WGS/WES 分析流程,但ilus不执行流程的具体步骤。你需要自己手动投递任务,不过执行过程不再依赖ilus,这也是为何我称之为半自动化的原因(这其实是一个优点,下文我会说到)。虽然如此,但 ilus 会帮你将最重要的流程和分析步骤生成出来,你只要按步骤投递就可以了。
目前 ilus 含有三个功能模块,分别是:
第一、
WGS全基因组数据分析流程模块。这个模块基于 GATK 最佳实践,采用bwa-mem + GATK,这和我之前的 WGS 系列教程是相互呼应的,但在 ilus 中被我做的更好用了。这个模块集成了比对、排序、同一个样本多lane 数据合并、标记重复序列(Markduplicates)、HaplotypeCaller gvcf 生成、多样本联合变异检测(Joint-calling)和变异质控(Variant quality score recalibrator, VQSR) 这 5 个完整的过程。而且这个模块同样适用于WES数据的分析,只需要将配置文件variant_calling_interval设置为WES 的外显子捕获区间即可(下文详述)。第二、
genotype-joint-calling联合变异检测模块。这个模块是从 ilus WGS 中分离出来的,目的是让我们可以从样本的 gvcf 文件 (注意 gvcf 文件和 VCF 文件是完全不同的)开始进行变异检测,这个功能很有用。特别是在碰到需要分多批次完成 WGS/WES 数据分析的时候(在大型基因组学项目中,这是很常见的),我们可以分批次生成 gvcf,最后再整理一个总的 gvcf 文件列表,然后用该模块完成后续步骤,这增加了分析流程的灵活度。第三、
VQSR变异质控模块,同样是从 ilus WGS 中分离出来的,目的是方便我们可以对变异结果进行独立的VQSR质控。
需要注意的是 ilus 不包含对原始 fastq 数据的质控,ilus 流程默认你输入的测序数据都是清洗好的 clean data。
ilus 不直接运行任务有如下两个方面的考虑:
- 第一,避免在 ilus 的核心功能之外做关于任务调度方面的优化。不同的计算集群(本地和云上),作业被调度的方式是多种多样的,如果将这些情况都一一考虑进去,ilus 会变得臃肿复杂,并且还不一定能够符合真实的需要,反而会导致一部分人无法有效使用 ilus,也容易在跟多系统任务管理缠斗的过程中丢失 ilus 真正要解决的问题。因此,作为一个轻量级的工具,我在设计 ilus 的时候从一开始就没将自动投递和运行任务的功能考虑在内。我更希望它作为一个框架程序,严格依据你的输入数据和配置文件的信息,生成符合你分析需求的流程脚本。
如果要实现作业的自动投递和监控,ilus 希望将来可以通过外部插件的形式来实现,但目前需要手动去投递这些按次序生成的脚本。
- 第二,增加流程灵活性和可操控性。ilus 所生成的流程都有一个特点,就是它们都将完全独立于 ilus,不会再依赖 ilus 的任何功能,这个时候即使你将 ilus 彻底从系统中卸载删除掉都没关系(这个特性是我有意而为的)。另外,ilus 所生成的可执行脚本(shell) 每一行都可以独立运行,彼此之间互不影响。
这样做的好处是,你可以按照计算机集群的特点将脚本拆分成若干个子脚本(如果你的样本不多或者集群资源不充足,也可以不拆分),然后再分别投递任务,这样可以大大加快任务的完成速度,这也是目前并行投递 ilus 任务的唯一方式,我也非常建议你应该这样去做(我的项目基本都是通过这样的方式来完成的)。
至于每一步要拆成多少个子脚本取决于你自己的具体情况。举个例子,比如你一共有 100 个样本,第一步的 xxx.step1.bwa.sh 比对脚本中将一共有 100 个比对命令,每一行都是一个样本的比对指令。由于这 100 个命令彼此独立互不依赖,因此你可以放心地将该步骤拆分为 100(或者任意小于 100 )个子脚本,然后再手动投递这些任务。
至于如何将一个完整的执行脚本拆分为多个,你既可以自己写程序完成,也可以使用我在 ilus 中提供的 yhbatch_slurm_jobs.py 程序来完成,但要注意,我提供的这个程序是基于 slurm 任务调度系统的(尚未测试过其它系统),不一定符合你的集群配置,但它的作用和意义我刚刚也做了说明,如果你觉得这个程序不能直接满足你的需求,你可以参照和修改。虽然 ilus 不做任务调度和执行,但是这个方法却能够增加流程控制的灵活性和操控性。
此外,当你有成千上万的样本需要分析时,实现对任务完成状态的监控是极其重要的一项任务。这个过程如果你打算手动来检查,那么一定是低效、易出错且极易让人发狂的。

我在 ilus 中充分考虑到了这一点,因此在生成流程的时候会为每个任务添加一个可识别的结束标记,我们只需要查看这个标记就行了(参考下文 WGS 的例子)。
有结束标志其实还不够,一旦我们的任务数量庞大,假如每次都要手动打开某些文件检查这个标志的话,那未免太过于麻烦。因此,我在 ilus 中还同时实现了一个专门用来检查任务作业完成状态的程序,具体用法同样参考下文 WGS 例子。
如何安装
ilus 是基于 Python 编写的,同时支持 Python3.7+ 和 Python2.7+,稳定版本的代码发布至 PyPI。因此要使用 ilus, 直接通过 pip 这个 Python 包管理工具进行安装即可,非常方便:
$ pip install ilus
该命令除了主程序 ilus 之外,还会自动将 ilus 所依赖的其它 Python 包自动装上。安装完成之后,在命令行中执行 ilus,如果能看到类似如下的内容,那么就说明安装成功了。
$ ilus
usage: ilus [-h] {WGS,genotype-joint-calling,VQSR} ...
ilus: error: too few arguments
ilus 的源代码托管在 github 中:https://github.com/ShujiaHuang/ilus
快速开始
执行 ilus --help 可以看到 WGS, genotype-joint-calling 和 VQSR 这三个功能模块。
$ ilus --help
usage: ilus [-h] {WGS,genotype-joint-calling,VQSR} ...
ilus: A WGS analysis pipeline.
optional arguments:
-h, --help show this help message and exit
ilus commands:
{WGS,genotype-joint-calling,VQSR}
WGS Creating pipeline for WGS(from fastq to genotype VCF)
genotype-joint-calling Genotype from GVCFs.
VQSR VQSR
下面,我通过例子逐一介绍如何使用这三个模块。
全基因组和全外显子数据分析
全基因组数据分析流程(WGS)的运行脚本通过 ilus WGS 来生成,用法如下:
$ ilus WGS --help
usage: ilus WGS [-h] -C SYSCONF -L FASTQLIST [-P WGS_PROCESSES]
[-n PROJECT_NAME] [-f] [-c] -O OUTDIR
optional arguments:
-h, --help show this help message and exit
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying details about
system.
-L FASTQLIST, --fastqlist FASTQLIST
Alignment FASTQ Index File.
-O OUTDIR, --outdir OUTDIR
A directory for output results.
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. Default value: test
-P WGS_PROCESSES, --Process WGS_PROCESSES
Specific one or more processes (separated by comma) of
WGS pipeline. Defualt value:
align,markdup,BQSR,gvcf,genotype,VQSR. Possible
values: {align,markdup,BQSR,gvcf,genotype,VQSR}
-f, --force_overwrite
Force overwrite existing shell scripts and folders.
-c, --cram Covert BAM to CRAM after BQSR and save alignment file storage.
其中,-C, -L 和 -O 是三个最重要且 必须的参数,其余的按照你的实际需要做选择。 -O 参数比较简单,就是一个输出目录,该目录如果不存在,无需担心,ilus 会自动创建。
最重要的是 -C 和 -L 这两个参数。 -C 参数是 ilus 的配置文件,如果没有这个文件 ilus 就无法正确生成分析流程了,因此它十分重要;-L 是输入文件,这个文件的格式有固定要求,一共 5 列,每一列都是流程所必须的信息。
下面,我分别对这两个文件的格式展开说明。
首先是 -C 配置文件,你需要在文件中填写好分析流程所需的所有程序路径、GATK bundle 文件路径、参考基因组 fasta 文件的路径以及各个关键步骤所对应的参数。
需要注意的是 bwa mem 的比对索引文件前缀要与配置文件的 resources.reference 的名字相同,并放在同一个文件夹里。如下:
/path/human_reference/GRCh38/
|-- human_GRCh38.fa
|-- human_GRCh38.dict
|-- human_GRCh38.fa.amb
|-- human_GRCh38.fa.ann
|-- human_GRCh38.fa.bwt
|-- human_GRCh38.fa.fai
|-- human_GRCh38.fa.pac
`-- human_GRCh38.fa.sa
human_GRCh38.fa 是 resources.reference,后面 6 份以 human_GRCh38.fa 为前缀的是 bwa mem 所需的比对索引文件,human_GRCh38.dict 也是一份必须的文件。配置文件要使用 Yaml 语法 来编写,这里我提供一份 配置文件的模板,大家可以直接参考:
aligner:
bwa: /path/to/BioSoftware/local/bin/bwa
bwamem_options: [-Y -M -t 8]
samtools:
samtools: /path/to/BioSoftware/local/bin/samtools
sort_options: ["-@ 8"]
merge_options: ["-@ 8 -f"]
stats_options: ["-@ 8"]
bcftools:
bcftools: /path/to/BioSoftware/local/bin/bcftools
concat_options: ["-a --rm-dups all"]
bedtools:
bedtools: /path/to/BioSoftware/local/bin/bedtools
genomecov_options: ["-bga -split"]
sambamba:
sambamba: /path/to/BioSoftware/local/bin/sambamba
sort_options: ["-t 8"]
merge_options: ["-t 8"]
markdup_options: []
verifyBamID2:
# Link of VerifyBamID2: https://github.com/Griffan/VerifyBamID
verifyBamID2: /path/to/BioSoftware/local/bin/verifyBamID2
options: [
# Download from: https://github.com/Griffan/VerifyBamID/tree/master/resource
"--SVDPrefix /path/to/BioSoftware/verifyBamID2/1.0.6/resource/1000g.phase3.10k.b38.vcf.gz.dat"
]
bgzip: /path/to/BioSoftware/local/bin/bgzip
tabix: /path/to/BioSoftware/local/bin/tabix
gatk:
gatk: /path/to/BioSoftware/gatk/4.1.4.1/gatk
markdup_java_options: ["-Xmx10G", "-Djava.io.tmpdir=/your_path/cache"]
bqsr_java_options: ["-Xmx8G", "-Djava.io.tmpdir=/your_path/cache"]
hc_gvcf_java_options: ["-Xmx4G"]
genotype_java_options: ["-Xmx8G"]
vqsr_java_options: ["-Xmx10G"]
CollectAlignmentSummaryMetrics_jave_options: ["-Xmx10G"]
# Adapter sequencing of BGISEQ-500. If you use illumina(or other) sequencing system you should
# change the value of this parameter.
CollectAlignmentSummaryMetrics_options: [
"--ADAPTER_SEQUENCE AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA",
"--ADAPTER_SEQUENCE AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG"
]
genomicsDBImport_options: ["--reader-threads 12"]
use_genomicsDBImport: false # Do not use genomicsDBImport to combine GVCFs by default
vqsr_options: [
"-an DP -an QD -an FS -an SOR -an ReadPosRankSum -an MQRankSum -an InbreedingCoeff",
"-tranche 100.0 -tranche 99.9 -tranche 99.5 -tranche 99.0 -tranche 95.0 -tranche 90.0",
"--max-gaussians 6"
]
# ``interval`` for create gvcf. The value could be a interval region file in bed format or could be a list here
interval: ["chr1", "chr2", "chr3", "chr4", "chr5", "chr6", "chr7", "chr8", "chr9",
"chr10", "chr11", "chr12", "chr13", "chr14", "chr15", "chr16", "chr17",
"chr18", "chr19", "chr20", "chr21", "chr22", "chrX", "chrY", "chrM"]
# Specific variant calling intervals.
# The value could be a file in bed format (I show you a example bellow) or a interval of list.
# Bed format of interval file only contain three columns: ``Sequencing ID``, ``region start`` and ``region end``,e.g.:
# chr1 10001 207666
# chr1 257667 297968
# These invertals could be any regions alone the genome as you wish or just set the same as ``interval`` parameter above.
variant_calling_interval: ["chr1", "chr2", "chr3", "chr4", "chr5", "chr6", "chr7", "chr8", "chr9",
"chr10", "chr11", "chr12", "chr13", "chr14", "chr15", "chr16", "chr17",
"chr18", "chr19", "chr20", "chr21", "chr22", "chrX", "chrY", "chrM"]
# variant_calling_interval: ["./wgs_calling_regions.GRCh38.interval.bed"]
# GATK bundle
bundle:
hapmap: /path/to/BioDatahub/gatk/bundle/hg38/hapmap_3.3.hg38.vcf.gz
omni: /path/to/BioDatahub/gatk/bundle/hg38/1000G_omni2.5.hg38.vcf.gz
1000G: /path/to/BioDatahub/gatk/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz
mills: /path/to/BioDatahub/gatk/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
1000G_known_indel: /path/to/BioDatahub/gatk/bundle/hg38/Homo_sapiens_assembly38.known_indels.vcf.gz
dbsnp: /path/to/BioDatahub/gatk/bundle/hg38/Homo_sapiens_assembly38.dbsnp138.vcf.gz
# Set Reference
resources:
reference: /path/to/BioDatahub/human_reference/GRCh38/human_GRCh38.fa
如果你不知道各个步骤最佳的参数是什么的话,不妨使用我配置模板中所提供的。
在配置文件中,bwa、samtools、bcftools、bedtools、gatk、bgzip 和 tabix 都是必须的生信软件,需要自行安装,然后再将路径填入到对应的参数里。
verifyBamID2 仅用于计算样本是否存在污染,它并不是一个必填的参数,如果你的配置文件中没有这个参数,则代表流程不对样本的污染情况进行推算,如果有那么你要自行安装并下载与之配套的 resource 数据,模板里我也告诉你该去哪里下载相关的数据了(没注意的话,建议回头去看看上面的配置模板信息)。
还有,配置文件 GATK 参数中的 Djava.io.tmpdir 缓存文件路径记得重新设置为你自己的文件夹路径。
此外,要注意配置文件中的 variant_calling_interval 参数。这是一个专门用来指定变异检测区间的参数。在以上配置例子里,我给出了从 chr1 到 chrM 这 25 条染色体,意思就是告诉流程只对这 25 条染色体做变异检测。如果你在参数里只列出一条染色体,或者仅仅给出一个染色体区间(区间信息按照 chrom:start-end 格式),比如 chr1:1-10000,那么 ilus 将只在你给定的这个区间里做变异检测。
这是一个非常灵活有用的参数,因为 variant_calling_interval 可以任意指定的,除了可以按照我例子给出的赋值方式之外,你还可以将区间 文件的路径 赋给这个参数。我们知道 WGS 和 WES 有很多步骤是完全相同的,只在变异检测的区间上存在差别——WES数据没有必要也不能 在全染色体上做变异检测,它只在外显子捕获区域里进行。
这个时候你只需要将外显子捕获区域的文件——注意是文件,这个文件的内容可以是 .interval_list 格式、.list 格式、.intervals 格式或者 .bed 格式。其中,.list 格式和 .intervals文件格式必须如下所示:
chr1:63697-63697
chr1:101158-101158
chr1:103241-103241
chr1:104108-104108
chr1:185336-185336
chr1:261495-261495
chr1:598862-598862
chr1:601606-601606
chr1:700596-700596
chr1:725086-725086
而 .interval_list 格式和 .bed 格式参照 GATK的说明,你不需要手动拆分成一个个的区间,只需将这个文件的路径赋给这个参数就可以了,这时流程就成了 WES 分析流程。这也是为何 ilus 同时是一个WGS 和 WES 分析流程生成器的原因。
另外,ilus 必需的公用数据集是:gatk bundle 和基因组参考序列(fasta 文件)。
【注意】如果你项目的样本量少于 10 那么 GATK 将不计算
InbreedingCoeff的值,此时配置文件中vqsr_options不需要设置-an InbreedingCoeff,可以将其去掉。
接下来是由 -L 参数指定的输入文件,文件里包含了 WGS/WES 分析流程所必需的一切信息。这是一个需要你自己来准备的文件,文件各列的格式信息如下:
- [1] SAMPLE,样本名;
- [2] RGID,Read Group,使用
bwa mem时通过 -R 参数指定的read group; - [3] FASTQ1,Fastq1 文件的路径;
- [4] FASTQ2,Fastq2 文件路径,如果是Single End测序,没有fastq2的话,该列用空格代替;
- [5] LANE,fastq 的 lane 编号。
这五个信息中
RGID要求严格,最容易出错。RGID一定要设置正确(正确的编写方式参考下面的例子或者这篇文章),否则你流程会出错。
另外,假如某些样本有多个 lane 的测序数据,或者同一个 lane 的数据被拆分成了很多个子文件,这个时候也不需要手动合并这些 fastq 数据,只需要依照 -L 的格式要求编写在输入文件里即可。对于这些同属一个样本的数据,ilus 都会根据样本 ID 在生成的流程中自动帮你合并。
下面我给出一个 -L 输入文件的例子,其中样本 HG002, HG003和 HG004 的数据就有分拆的情况(哪怕碎成一万份也没问题):
#SAMPLE RGID FASTQ1 FASTQ2 LANE
HG002 "@RG\tID:CL100076190_L01\tPL:COMPLETE\tPU:CL100076190_L01_HG002\tLB:CL100076190_L01\tSM:HG002" /path/to/NA24385_CL100076190_L01_read_1.clean.fq.gz /path/to/NA24385_CL100076190_L01_read_2.clean.fq.gz CL100076190_L01
HG002 "@RG\tID:CL100076190_L02\tPL:COMPLETE\tPU:CL100076190_L02_HG002\tLB:CL100076190_L02\tSM:HG002" /path/to/NA24385_CL100076190_L02_read_1.clean.fq.gz /path/to/NA24385_CL100076190_L02_read_2.clean.fq.gz CL100076190_L02
HG003 "@RG\tID:CL100076246_L01\tPL:COMPLETE\tPU:CL100076246_L01_HG003\tLB:CL100076246_L01\tSM:HG003" /path/to/NA24149_CL100076246_L01_read_1.clean.fq.gz /path/to/NA24149_CL100076246_L01_read_2.clean.fq.gz CL100076246_L01
HG003 "@RG\tID:CL100076246_L02\tPL:COMPLETE\tPU:CL100076246_L02_HG003\tLB:CL100076246_L02\tSM:HG003" /path/to/NA24149_CL100076246_L02_read_1.clean.fq.gz /path/to/NA24149_CL100076246_L02_read_2.clean.fq.gz CL100076246_L02
HG004 "@RG\tID:CL100076266_L01\tPL:COMPLETE\tPU:CL100076266_L01_HG004\tLB:CL100076266_L01\tSM:HG004" /path/to/NA24143_CL100076266_L01_read_1.clean.fq.gz /path/to/NA24143_CL100076266_L01_read_2.clean.fq.gz CL100076266_L01
HG004 "@RG\tID:CL100076266_L02\tPL:COMPLETE\tPU:CL100076266_L02_HG004\tLB:CL100076266_L02\tSM:HG004" /path/to/NA24143_CL100076266_L02_read_1.clean.fq.gz /path/to/NA24143_CL100076266_L02_read_2.clean.fq.gz CL100076266_L02
HG005 "@RG\tID:CL100076244_L01\tPL:COMPLETE\tPU:CL100076244_L01_HG005\tLB:CL100076244_L01\tSM:HG005" /path/to/NA24631_CL100076244_L01_read_1.clean.fq.gz /path/to/NA24631_CL100076244_L01_read_2.clean.fq.gz CL100076244_L01
接下来,举例说明 ilus WGS 的使用和整个流程的结构。
例子1:从头开始生成 WGS 分析流程
$ ilus WGS -c -n my_wgs_project -C ilus_sys.yaml -L input.list -O output/
这个命令的意思是,项目 (-n) my_wgs_project 依据配置文件 (-C) ilus_sys.yaml
和输入数据 (-L )input.list 在输出目录 output 中生成一个 WGS
分析流程。同时流程在完成分析之后将 BAM 比对文件自动转为 (-c) CRAM。CRAM 和 BAM 格式和可被操作的方式几乎相同,但可以节省大约三分之一的空间,如果不设置 -c 参数,则保留原来的 BAM 文件。
以上命令顺利运行之后,输出目录 output 里会生成如下 4 个文件夹:
00.shell/
01.alignment/
02.gvcf/
03.genotype/
它们分别用于存放流程产生的各类不同数据,其中:
00.shell流程shell脚本的汇集目录;01.alignment以样本为单位存放比对结果;02.gvcf存放各个样本的gvcf结果;03.genotype存放最后变异检测的结果。
00.shell 目录里有分析流程执行脚本和日志目录,我们要投递和执行脚本都在这:
/00.shell
├── loginfo
│ ├── 01.alignment
│ ├── 01.alignment.e.log.list
│ ├── 01.alignment.o.log.list
│ ├── 02.markdup
│ ├── 02.markdup.e.log.list
│ ├── 02.markdup.o.log.list
│ ├── 03.BQSR
│ ├── 03.BQSR.e.log.list
│ ├── 03.BQSR.o.log.list
│ ├── 04.gvcf
│ ├── 04.gvcf.e.log.list
│ ├── 04.gvcf.o.log.list
│ ├── 05.genotype
│ ├── 05.genotype.e.log.list
│ ├── 05.genotype.o.log.list
│ ├── 06.VQSR
│ ├── 06.VQSR.e.log.list
│ └── 06.VQSR.o.log.list
├── my_wgs_project.step1.bwa.sh
├── my_wgs_project.step2.markdup.sh
├── my_wgs_project.step3.bqsr.sh
├── my_wgs_project.step4.gvcf.sh
├── my_wgs_project.step5.genotype.sh
└── my_wgs_project.step6.VQSR.sh
投递任务运行流程时,按顺序从 step1 依次执行到 step6 即可。loginfo/ 文件夹下记录了各个样本所有步骤的运行状态,你可以通过检查各个任务的 .o.log.list 日志文件,获得每个样本是否都有成功结束的标记。
如果成功了,你将在日志文件的末尾看到一句类似于 [xxxx] xxxx done 的标记。用我在 ilus 中提供的程序 check_jobs_status.py,你就可以很方便地知道哪些已经顺利完成,哪些还没有。这个脚本会帮你将那些未完成的任务全部输出,方便检查问题或重新执行这部分未完成的任务。
check_jobs_status 的用法如下:
$ python check_jobs_status.py loginfo/01.alignment.o.log.list > bwa.unfinish.list
如果这个 bwa.unfinish.list 文件是空的,并输出了** All Jobs done **,那么就代表你 step1 的所有任务都成功结束了,以此类推。如果有未完成的,那么你要提取出来,重新投递。
例子2:只生成 WGS 流程中的某个/某些步骤
有时,我们并不打算(或者没有必要)从头到尾完整地将 WGS 流程执行下去,比如我们只想完成从 fastq 比对到生成 gvcf 结束,暂时不想执行 genotype 和 VQSR——这情况在大型基因组项目中很普遍,这时该怎么办呢?ilus 的 -P 参数就可以实现这个目的:
$ ilus WGS -c -n my_wgs_project -C ilus_sys.yaml -L input.list -P align,markdup,BQSR,gvcf -O output/
这样就只生成从 bwa 到 gvcf 的执行脚本,这对于需要分批次完成分析的项目来说是很有用的。而且 ilus 所输出的结果是以样本为文件夹作区分的,因此在相同的输出目录下,只要样本编号是不同的,那么不同批次的数据就不会存在相互覆盖的问题。
除此之外,-P 参数还有一个用途,那就是万一某个 WGS 步骤跑错了,需要重跑,那你就可以用 -P重跑特定的步骤。比如我需要重新生成 BQSR 这个步骤的运行脚本,那么就可以这样做:
$ ilus WGS -c -n my_wgs_project -C ilus_sys.yaml -L input.list -P BQSR -O ./output
不过,要特别注意,ilus 为了节省项目对存储空间的消耗,只会为每一个样本保留 BQSR 之后的总 BAM/CRAM 文件。因此,如果你要重新跑 BQSR 那就需要先确保 BQSR 的前一步(即,markdup)的 BAM 文件还没有被被删除。如果你在项目中一直使用的都是 ilus 那么是不用担心这个问题的,因为 ilus 执行任务时具有 原子属性,也就是说只有当步骤中所有过程都成功结束了才会将那些完全不需要的文件删除掉。所以,如果 ilus 的 BQSR 没有正常结束,那么前一步 markdup 的 BAM 文件是会被完整保留住的。
目前
-P参数能指定的分析模块必须属于「align,markdup,BQSR,gvcf,genotype,VQSR」中的一个或多个,并用英文逗号隔开,中间不可以有空格。
genotype-joint-calling
如果我们已经有了各个样本的 gvcf 数据,现在要用这些 gvcf 完成多样本的联合变异检测(Joint-calling),那么就要用 genotype-joint-calling 了。具体用法如下:
$ ilus genotype-joint-calling --help
usage: ilus genotype-joint-calling [-h] -C SYSCONF -L GVCFLIST
[-n PROJECT_NAME] [--as_pipe_shell_order]
[-f] -O OUTDIR
optional arguments:
-h, --help show this help message and exit
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying details about
system.
-L GVCFLIST, --gvcflist GVCFLIST
GVCFs file list. One gvcf_file per-row and the format
should looks like: [interval gvcf_file_path]. Column
[1] is a symbol which could represent the genome
region of the gvcf_file and column [2] should be the
path.
-O OUTDIR, --outdir OUTDIR
A directory for output results.
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. [test]
--as_pipe_shell_order
Keep the shell name as the order of `WGS`.
-f, --force Force overwrite existing shell scripts and folders.
-L 是 genotype-joint-calling 的输入参数,它要一个 gvcf list 文件,这个文件很简单,由两列构成:第一列是每个 gvcf 文件所对应的区间或者染色体编号;第二列是这份 gvcf 文件的路径。目前 ilus 要求各个样本的 gvcf 都按照主要染色体(1-22、X、Y、M)分开,举个例子:
$ ilus genotype-joint-calling -n my_project -C ilus_sys.yaml -L gvcf.list -O genotype --as_pipe_shell_order
其中 gvcf.list 的格式如下:
chr1 /path/sample1.chr1.g.vcf.gz
chr1 /paht/sample2.chr1.g.vcf.gz
chr2 /path/sample1.chr2.g.vcf.gz
chr2 /path/sample2.chr2.g.vcf.gz
...
chrM /path/sample1.chrM.g.vcf.gz
chrM /path/sample2.chrM.g.vcf.gz
这个例子里 gvcf.list 只有两个样本(文件顺序随意)。参数 --as_pipe_shell_order 可加也可不加(默认是不加,但我建议加),它唯一的作用就是按照 ilus WGS 流程的方式输出执行脚本的名字,维持和 WGS 流程一样的次序和相同的输出结构。
VQSR
这个模块只用来生成基于 GATK VQSR 的变异质控执行脚本。VQSR 的大致原理我以前在我的知识星球 (达尔文生信星球) 中简要回答过。
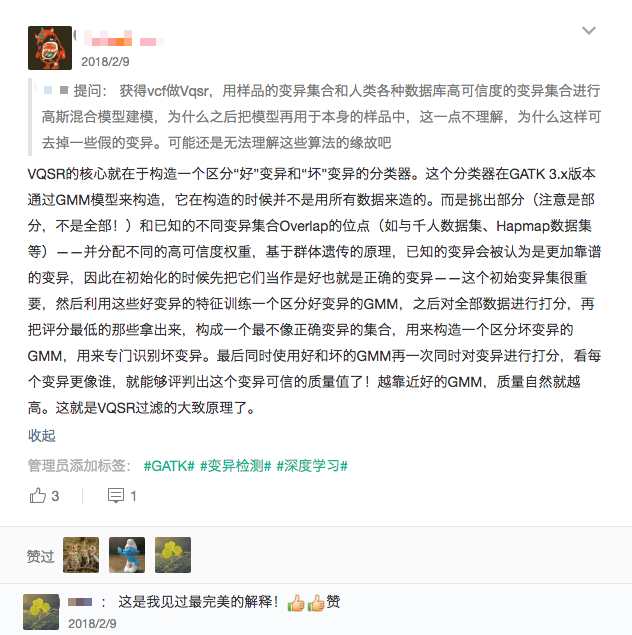
我们如果已经有了最终的变异检测(VCF格式)结果,现在只想用 GATK VQSR 对这个变异数据做质控,那么就可以使用这个模块了,用法和 genotype-joint-calling 大同小异,如下:
$ ilus VQSR --help
usage: ilus VQSR [-h] -C SYSCONF -L VCFLIST [-n PROJECT_NAME]
[--as_pipe_shell_order] [-f] -O OUTDIR
optional arguments:
-h, --help show this help message and exit
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying details about
system.
-L VCFLIST, --vcflist VCFLIST
VCFs file list. One vcf_file per-row and the format
should looks like: [interval vcf_file_path]. Column
[1] is a symbol which could represent the genome
region of the vcf_file and column [2] should be the
path.
-O OUTDIR, --outdir OUTDIR
A directory for output results.
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. [test]
--as_pipe_shell_order
Keep the shell name as the order of `WGS`.
-f, --force Force overwrite existing shell scripts and folders.
跟 genotype-joint-calling 相比不同的是,ilus VQSR 的输入文件是 VCF 文件列表,并且 每行只有一个 VCF 文件路径,例子如下:
/path/chr1.vcf.gz
/path/chr2.vcf.gz
...
/path/chrM.vcf.gz
其它参数与 genotype-joint-calling 相同。文件列表中的 vcf 不需要事先合并,ilus VQSR 会帮你合并(如你已经合并成一份了,也一样使用),但你不要将两个或者多个不同项目的 VCF 混在一起,那完全不是一回事,必须得是同一个项目的。以下提供一个完整的例子:
$ ilus VQSR -C ilus_sys.yaml -L vcf.list -O genotype --as_pipe_shell_order
其它
关于文中我提到的 yhbatch_slurm_jobs.py 和 check_jobs_status.py 都在 ilus github 代码仓库的 scripts 目录里:
以上,就是关于 WGS/WES 流程生成器 ilus 的完整介绍,希望在有需要的时候它能成为你的一把小刀子,辅助你解决问题,使用过程中有任何问题都可以告知我或者在 github 上提给我。
订阅
文章首发于我的个人公众号:helixminer(碱基矿工)
